Generative Molecular Design Isn't As Easy As People Make It Look

I was taken aback by a recent CNBC article entitled “Generative AI will be designing new drugs all on its own in the near future”. I should know better than to pay attention to AI articles in the popular press, but I feel that even scientists working in drug discovery may have a skewed perception of what generative AI can and can’t do. To understand exactly what’s involved, it might be instructive to walk through a typical generative molecular design workflow and point out a few things. First, these programs are far from autonomous. Even when presented with a well-defined problem, generative algorithms produce a tremendous amount of nonsense. Second, domain expertise is essential when sifting through the molecules produced by a generative algorithm. Without a significant medicinal chemistry background, one can’t make sense of the results. Third, while a few nuggets exist in the generative modeling output, a lot of work and good old-fashioned cheminformatics are required to extract them.
For this particular exercise, I examined the output of DiffLinker, a program recently published by Ilia Igashov and colleagues. DiffLinker uses a diffusion model to generate the chemical structures of linkers that can connect two or more chemical fragments. Methods like this have a range of applications in drug discovery, including fragment-based design, PROTAC optimization, and the replacement of peptide backbones. One of the exciting aspects of DiffLinker is its ability to work in 3D. Unlike some other generative models that produce 1D structures as SMILES and convert them to 3D for scoring, DiffLinker operates directly on 3D structures. In addition, DiffLinker can perform fragment linking in the context of a protein binding site and avoid steric clashes with the protein if the binding site is provided. The input to DiffLinker is a set of chemical fragments and attachment atoms. This is illustrated in the figure below, which shows the experimental structures of two fragments bound to HSP90, a molecular chaperone implicated in tumor growth. The two atoms highlighted in magenta are the atoms that DiffLinker will connect.

The Bonds That Keep Us Together
It’s important to remember that DiffLinker doesn’t operate on molecules but on a point cloud. This point cloud is simply a set of points in space where each point has an associated atom type. The diffusion model uses distributions learned from experimental crystal structures to fit a set of points with defined atom types. As such, DiffLinker doesn’t have a notion of bonds or bond orders. Its output is a set of x,y,z coordinates and element types. To be used with any molecular modeling software, the output from DiffLinker must undergo a conversion that involves assigning bonds and bond orders. This is harder than it might appear. The assignment of bonds is relatively easy. We can loop over all pairs of atoms and assign a bond between any two atoms where the interatomic distance is less than the sum of the covalent radii plus some tolerance (typically 0.4-0.5 Å). Assigning bond orders, on the other hand, is tricky. At first, this seems straightforward: bond lengths are shorter for double and triple bonds. We should be able to create a lookup table that relates bond length to bond order and assign the bond orders. That’s great until we consider conjugated pi systems with alternating single and double bonds. Given the structure on the left below, going to the structure on the right is not trivial. A 2001 whitepaper by Roger Sayle provides a more complete description of the problem of assigning bonds and bond orders.

Of course, the astute reader now thinks, “We have trusty open source Cheminformatics toolkits that can solve this problem, right?” Unfortunately, the answer here is “not really.” Both the RDKit and Open Babel have routines for assigning bonds and bond orders. However, these routines were primarily designed to process the output of quantum chemical calculations with hydrogen atoms attached to the corresponding heavy atoms. If we give the RDKit an XYZ file from DiffLinker, it makes a fine mess of things.

It’s not that the routines in the RDKit are wrong; it’s just that this is not the task they were designed to perform. OpenBabel does a bit better but still has some problems. Here’s OpenBabel’s conversion of the same XYZ file.

Seeing Double
When running DiffLinker, users can specify the number of solutions to generate. Since I wanted several choices, I specified 1,000 output molecules using the –output flag. The OEChem workflow above enabled me to parse the output from DiffLinker and generate mostly correct output. The first thing I wanted to do was remove duplicate ideas. I generated an InChI key for each molecule and counted the duplicates to do this. I could have also identified duplicates using isomeric SMILES, but the InChI key normalizes tautomers and can provide a more robust approach. The table below shows the InChI key for the 10 most frequently occurring molecules (don’t worry, we’ll look at structures soon) and the number of times the structure was generated.

As we can see from the table, one structure was generated 145 times. Of course, comparing the InChI keys only tells us that the same molecular graph was generated multiple times. It’s possible that the same molecule could assume different poses and/or conformations in the binding site. One way to investigate this is to calculate the pairwise root mean squared deviation (RMSD) between all pairs and examine the RMSD distribution. If we do this, we end up with the distribution plotted below. As we can see from the plot, the RMSD values are less than 0.3 A, indicating that all copies have similar conformations and poses.

To confirm the numbers, I wrote out the structures of all 145 copies and visualized them in PyMol. Indeed, the poses and conformations are the same. The astute reader may notice that the molecule below isn’t chemically stable; we’ll get to that later.
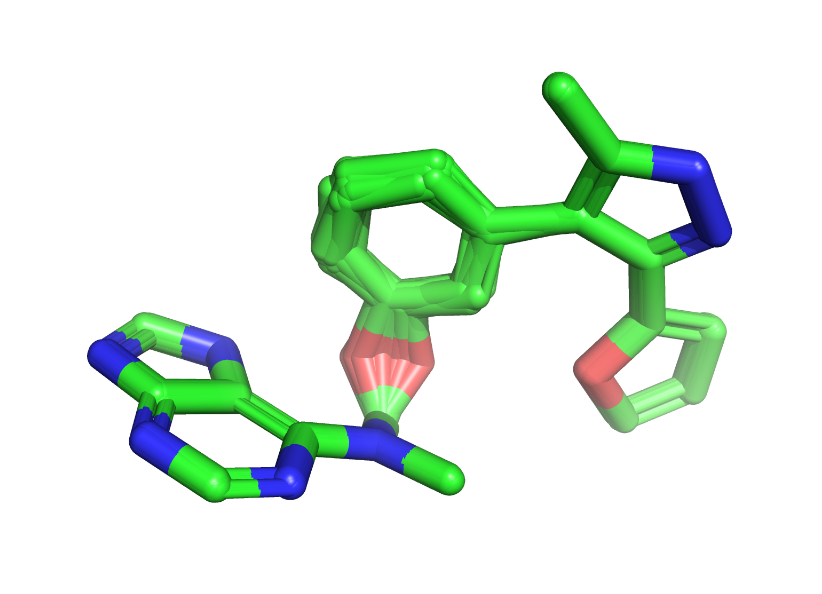
After a few more spot checks, I felt comfortable only keeping one copy of the duplicate structures. Since the data is stored in a Pandas dataframe, it’s easy to use the drop_duplicates method to remove rows with the same InChI key from the table. After removing duplicates, we have 581 unique molecules remaining.
Put a Ring On It
One troubling aspect of molecule generation algorithms is their tendency to produce molecules containing ring systems that aren’t chemically stable. This instability can sometimes be attributed to ring strain. However, in many cases, it’s due to chemically unstable bonds or partially substituted aromatic systems that are likely to oxidize to more stable aromatic forms. It would be difficult to create rules for assessing the stability of every crazy ring system dreamed up by a generative model. Instead of trying to be overly clever, I devised a simple approach, which I described in detail in a previous post. In brief, I wrote an algorithm to extract ring systems from a molecule. Once extracted, each ring system is compared with a lookup table containing all the ring systems in the ChEMBL database and their frequencies. If a ring system doesn’t occur in ChEMBL, it’s probably either unstable or difficult to synthesize. In either case, it will probably not be interesting to a medicinal chemist.
To evaluate the ring systems in the 537 molecules remaining after duplicate removal, I used the RingSystemLookup class in the useful_rdkit_utils toolkit. 144 of the generated molecules contained ring systems that occurred fewer than 100 times in ChEMBL. The figure below shows a few examples of ring systems that do not occur in ChEMBL. Even a cursory examination shows that these are not molecules anyone could synthesize. One may ask whether requiring a ring system to appear 100 times in ChEMBL to be considered valid is an overly strict criterion. On the contrary, as we will see below, 100 may not have been strict enough.

Filters, Filters, Filters
As I started to look through the DiffLinker results, I noticed that many of the molecules contained chemically unstable functionality. I wanted to identify and remove these molecules, so I turned to the REOS (Rapid Elimination of Swill) functionality in the useful_rdkit_utils toolkit. REOS has several collections of functional groups that are problematic in drug discovery. The collection includes moieties known as reactive, toxic, or interfering with biological assays. The REOS class has several filter sets available. The “Dundee” filter set feels most familiar to me and has become my default. Following the application of the Dundee filters to the remaining 537 molecules, only 143 met the criteria and were retained. The table below shows the rules that eliminated the largest number of molecules.

130 molecules were removed due to the ‘het-C-het’ rule which represents the SMARTS pattern “[NX3R0,NX4R0,OR0,SX2R0][CX4][NX3R0,NX4R0,OR0,SX2R0]”. This pattern matches an acyclic aliphatic carbon flanked by two heteroatoms in functional groups such as acetals, ketals, aminals, hemiaminals, and diols These functional groups tend to hydrolyze under acidic conditions and may lead to decomposition. The figure below shows a few examples with the ‘het-C-het’ functionality highlighted.

Then again, this filtering is probably too aggressive. In a 2021 paper in J Med Chem, Wu and Meanwell point out that not all het-C-het functionality is unstable. They mention that more than 90 marketed drugs contain these linkages and highlight several strategies for stabilizing these bonds. As with many of the strategies detailed herein, it’s complicated.
A Strain on Our Relationship
Using the filters described above, I winnowed the original list of 1,000 molecules to 143. As I examined the remaining molecules, I noticed several geometric errors where bond lengths or angles were not standard, leading to highly strained structures. One example is shown below, where aromatic ring substituents are out of plane, and the bond angles coming off the ring are far from the preferred 120 degrees.
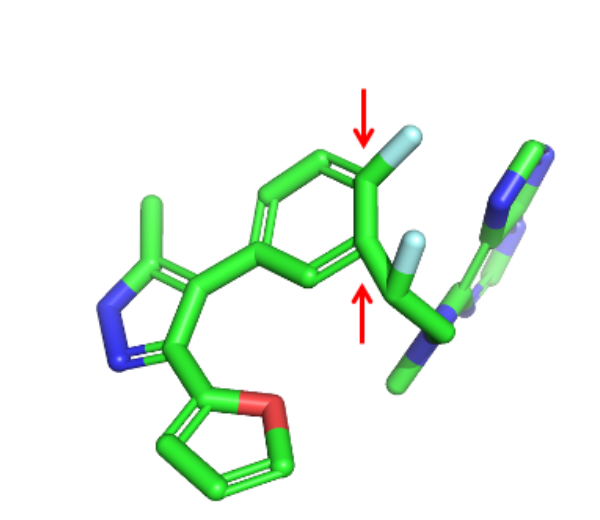
Fortunately, there is an open source software library for identifying these sorts of irregularities. The PoseBusters software published by Martin Buttenschoen and colleagues has 19 tests for structural errors, including bond lengths, bond angles, internal steric clashes, and clashes between protein and ligand. Running the PoseBusters tests on the remaining 143 molecules eliminated another 59, leaving 88 that passed all the filters. In addition to PoseBusters, filtering the structures for torsional strain would probably be a good idea. This task can be easily accomplished with software available in the open source MayaChem Tools suite.
Where Does This Leave Us?
At this point, it was relatively easy to look through the remaining 88 molecules to determine which might be followed up experimentally. However, as I looked at the molecules that passed the previous filters, I questioned the 100 ChEMBL example threshold I set when using the ring system filter. The table below shows 8 of the remaining 88 molecules with the lowest ChEMBL ring system frequencies. The number below the structures indicates the frequency of the linker ring system in ChEMBL. Many ring systems that appear fewer than 500 times aren’t stable. Surprisingly, cyclobutadiene, a classic Diels-Alder adduct, is found in 135 ChEMBL molecules. I’m also not crazy about the saturated heterocycle and partially saturated aromatic linkers highlighted below.

At the end of the day, two interesting themes emerged from this investigation. Both were simple motifs that coupled a 5 or 6-membered heterocycle with a short acyclic linker. Two examples are shown in the figure below. To assess the stability of the DiffLinker conformations, I generated 50 conformers of each molecule using the ETKDG method implemented in the RDKit. Each conformer was minimized with the MMFF forcefield and overlaid with the corresponding DiffLinker structure. The figure below shows an overlay of the confomer with the lowest RMSD. The DiffLinker structure is shown in green, with the closest low-energy conformer superimposed in blue. As we can see, even after PoseBusters filtering, the structures were strained. The green structure on the right contains an obviously eclipsed single bond, which doesn’t look happy. If this were a “real” project, I’d move to physics-based methods and perform molecular dynamics simulations to examine the stability of the ligand poses in the binding site.

Through this long-winded exercise, I’ve attempted to demonstrate that generative molecular design isn’t a seamless process. We don’t simply point a generative model at a protein binding site and wait for drugs to pop out. A great deal of domain expertise and patience is required to sift through generative modeling output and identify promising molecules for synthesis. Even after the work described above, there’s still more to do. Note that we didn’t integrate any sort of synthesizability assessment in this process. While there are automated approaches to designing organic syntheses, these tools have yet to achieve mainstream acceptance. Ultimately, the best arbiter for how to synthesize a molecule is an experienced organic chemist.
The filtering workflow above provides one method of processing the output from a generative model. An alternate approach is using human-in-the-loop machine learning to develop models that differentiate between desirable and undesirable molecules. In a 2023 paper, a group from Microsoft and Novartis described MolSkill, an approach that uses human feedback to model the drug-likeness of molecules. As part of this exercise, 35 Novartis medicinal chemists were asked to rank pairs of molecules and select the one they preferred. This input was subsequently used to train a machine learning model to recognize desirable molecule features. I wrote a 2023 post about MolSkill for those interested in learning more.
ACD 0-1-2-3..4-5
In 2022, my colleagues and I published a paper in J Med Chem that defined a framework for Automated Chemical Design (ACD) along two axes.
Ideas - did a person or machine come up with ideas for molecules?
Selections - did a person or a machine decide which molecules to synthesize?
We defined six ACD levels based on these dimensions, illustrated in the figure below.
0. Ideas and selections are defined by a person.
1. A machine provides ideas that are then selected by a person.
2. A person defines the available chemical space (e.g. reactions), and selections are made by a machine
3. A machine provides ideas which are also prioritized by a machine
4. The same as level 2, with multiple iterations.
5. The same as level 3, with multiple iterations

Much of what we see in the popular and scientific press makes it appear that generative models operate at ACD level 3 or beyond, but this isn’t the case. Like the examples detailed earlier in this post, most generative molecular design papers would be classified as ACD level 1. In those publications, an algorithm generates new molecules that meet some criteria, such as fitting to a binding site. A person then manually sorts through the generated molecules and decides which compounds to synthesize. Papers with titles like “Inhibitor of **Insert Name of Target** Discovered by Machine Learning” feel disingenuous when the final selections were driven by scientists with decades of drug discovery experience.
It’s important to note that the issues I’ve highlighted here aren’t exclusive to DiffLinker. Many of the generative algorithms I’ve experimented with exhibit similar pathologies. Generative AI can be useful, but it’s in its infancy. As one of my colleagues said, “There’s a lot of child-proofing that has to go on with these models.” While it’s possible to put up safety gates to remove some of the silly molecules these models produce, manual curation is still required. Gary Marcus said large language models (LLMs) “respond to corpus similarity, not concepts.” We need to think the same way about molecule generation. These models are not learning chemistry; they’re learning patterns in SMILES strings or points in space. While generative models provide an important source of ideas, we’re still a long way from algorithms that can design molecules on their own.
Acknowledgments
I'd like to thank Jacob Gora, Hakan Gunaydin, Ilia Igashov, Steven Kearnes, Mark Murcko, Patrick Riley, and Rebecca Swett for helpful discussions.
Code Availability
The code used in this analysis is available on GitHub https://github.com/PatWalters/practical_cheminformatics_posts/blob/main/generative_workflow/filter_difflinker.ipynb



Hi Pat, the set of structures starting with 135 all have the purine ring as less stable tautomer (repulsion between the lone pairs of N3 and N9). While it is not incorrect from a cheminformatic perspective to register a compound as the less stable tautomer, this will be an issue if the generated structures need to have the purine in this tautomeric form in order to be recognized by a target. I too would worry about a cyclopentadiene substructure (more from a general stability/reactivity perspective than for potential to function as a Diels Alder diene) but at least it’s not aromatic which will keep the PFI advocates happy.
ReplyDeleteThanks, PK. There's a lot more I could have talked about here. I have a couple of notebooks in Practical Cheminformatics Tutorials that deal with tautomer enumeration and selection
Deletehttps://github.com/PatWalters/practical_cheminformatics_tutorials/blob/main/fundamentals/stereo_and_tautomers.ipynb
https://github.com/PatWalters/practical_cheminformatics_tutorials/blob/main/qm/tautomer_energies.ipynb
Hi Pat,
ReplyDeleteThanks, it's a good article.
There are a couple of comments regarding the strain in the molecular structures.
Firstly, ligands need to change their structure upon binding, so small strains are normal.
Secondly, you cannot expect a model trained on such distorted, strained structures to produce relaxed structures. Quite the opposite, it may generate even more strained structures.
Therefore, I think that before checking the geometry of the structure, it should be relaxed.