For over two decades, many people,
including me, have been writing programs attempting to replicate "medicinal chemistry intuition". This ability to identify molecules that would be considered drug-like can be valuable in various areas, including
purchasing compounds for screening collections and prioritizing molecules output by generative algorithms or other denovo design methods. Currently, the most widely used approach for evaluating drug-likeness is the
QED method, published by Andrew Hopkins and coworkers at Pfizer in 2012. QED uses a weighted combination of calculated properties and structural alerts to generate a drug-likeness score for a molecule, with a higher score indicating a more drug-like molecule. Most recently, many generative molecular design methods have used QED as part of their objective function.
A new paper from scientists at Novartis and Microsoft presents an alternate approach, called MolSkill, for quantifying drug-likeness. In this study, 35 medicinal chemists at Novartis were presented with pairs of molecules and asked which of the two they preferred. The chemists' preferences were then used to train a machine learning model whose output was a score that predicted the preference for a molecule. A more negative score indicated a more highly preferred molecule. I was intrigued by the approach, so I decided to try it. As usual, the code and data used in this post are available on GitHub. To evaluate how the model performed, I calculated QED and MolSkill scores for four sets of molecules.
Marketed Drugs - 1935 marketed drugs taken from the ChEMBL database. These are drugs, so I'm considering this the gold standard.
ChEMBL Molecules - a set of 2000 molecules selected randomly from the ChEMBL database. These molecules were curated from the medicinal chemistry literature, so it's safe to assume they are somewhat drug-like.
REOS Molecules - 2000 molecules from the ChEMBL database that failed a wide range of functional group filters. In this case, I used the BMS filters from the rd_filters package.
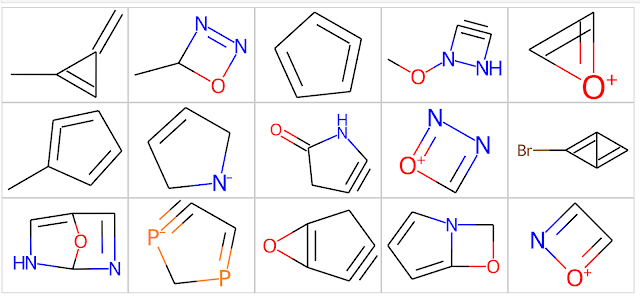
Odd Molecules - 2000 molecules that don't make a lot of sense. This requires a bit of explanation. The STONED SELFIES method is a simple generative modeling approach that works by permuting the SELFIES line notation for molecules. While the SELFIES generated by this approach obey standard valence rules, they're often the sorts of molecules that most organic chemists look at and say "that can't possibly exist". I generated these molecules by running the STONED SELFIES code on each molecule in the "ChEMBL Molecules" set above and generating up to 1000 analogs for each ChEMBL molecule. I then used the useful_rdkit_utils package to identify analogs with ring systems that do not occur in the ChEMBL database. This procedure generated over 5 million "odd molecules," from which I randomly selected 2000. Some examples from the "Odd Molecules" set are shown below. As you can see, they're odd.
The boxplot below shows the MolSkill scores for the four sets. As expected, the Drugs and ChEMBL datasets had scores that were more negative (better) than those of the REOS and Odd sets. It's interesting to note the median score for the ChEMBL dataset is lower than that of the Drug set. If we consider that the MolSkill model was trained on molecules from ChEMBL, this isn't particularly surprising. It's also interesting to note that although the MolSkill score doesn't explicitly encode objectionable functionality, it still assigns higher (worse) scores to the REOS and Odd sets.
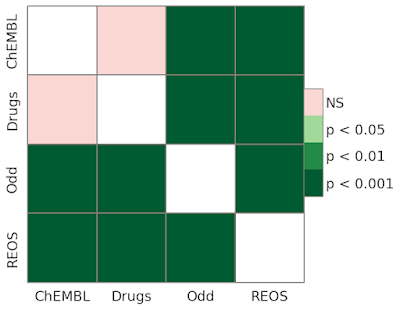
Of course, we want to be rigorous about our statistical analyses, so we'll use
scikit-posthocs to determine if the distributions above are different. Readers interested in learning more about post hoc tests and multiple comparisons may want to check out
this Practical Cheminformatics post. A quick examination of the heatmap below shows that all distribution differences can be considered significant at p < 0.001.
Those interested in hands-on experience with the MolSkill approach may want to try out an
interactive tool that David Huang from the Oloren AI team put together. Inspired by the MolSkill paper, David's tool displays molecules and asks the user to guess whether a molecule came from a pharmaceutical patent or a generative model.
So far so good; it looks like the MolSkill score does a good job of identifying drug-like molecules. However, I'd also like to see if this score is telling me something I can't learn from existing metrics like QED. Let's try the same exercise using the implementation of
QED in the RDKit. The boxplot below shows the distributions of QED for the same sets used above. In this case, the Drug and ChEMBL sets have QED distributions where the median is higher (better) than those of the REOS and Odd sets. QED explicitly encodes several undesirable functional groups that contribute negatively to the overall score, so unsurprisingly, the REOS set has the lowest median score.
As mentioned above, we'll use scikit-posthocs to examine the significance of the differences in the QED distributions. This time, we see that the difference between Drugs and ChEMBL is not significant, while the others are.
So where does this leave us? As mentioned above, I primarily use methods like these to remove molecules I don't want to purchase or synthesize. This becomes particularly relevant when considering the output of generative algorithms, which have an uncanny ability to generate strange molecules. The astute reader may ask, "why not simply use functional group filters like those in the rd_filters to remove the strange molecules?". Unfortunately, this doesn't work. Functional group filters are designed to identify functional groups like aldehydes and quinones that can be synthesized but may be reactive invivo or in an assay. Take a look at the chemical structures in the table above. Everything there is chemically unstable and unlikely to ever be synthesized. It's challenging to create filters for things that haven't existed in this universe.
However, like
Tom Robbins, I digress. Let's get back to the problem at hand. Would I use MolSkill over QED when selecting compounds? Probably not. In the MolSkill paper, the authors show that the two metrics are not correlated, but I wonder if MolSkill is telling me something I'm not getting from QED. At the end of the day, I like to use tools I understand. QED is based on simple descriptors I've used for years, and I know how it works. MolSkill is based on a neural network and is a bit of a black box.
While we're on this topic, I'd like to look at synthesizability, another approach to identifying odd molecules. Over the last few years, there has been a great deal of work on developing neural network models that can
propose synthetic routes to molecules. That's not what we're talking about here. In this case, we're not asking, "how do I synthesize this molecule?" we're asking, "has anyone ever synthesized a molecule like this?". An elegant approach to this question is the synthetic accessibility (SA) score published in
2009 by Peter Ertl and Ansgar Schuffenhauer. The SA score approach begins by using rules to derive a set of molecular fragments and their frequencies from almost a million molecules randomly selected from the PubChem database. The SA score for a new molecule can then be calculated by fragmenting the molecule according to the same set of rules and looking up the frequency of the fragments. If a molecule contains frequently occurring fragments, people have probably made similar molecules before, and we consider the molecule synthesizable. If the molecule contains rare fragments that haven't been seen before, it will likely be more challenging to synthesize. This description is a bit of an oversimplification. I'd urge interested readers to check out the original paper for the whole story.
Here are the SA scores for the same four sets of molecules used in the analyses above. With SA score, a lower value indicates a molecule that is more synthetically accessible. Interestingly, the Odd set sticks out like a sore thumb. This makes sense; the SA score penalizes molecules containing fragments not frequently found in the PubChem database. Ultimately, this comes down to "odd molecules contain odd fragments".
In the interest of completeness, we'll use scikit-posthocs and see that the differences between distributions are significant at p<0.001.
In this post, I've hopefully shown you that there are multiple ways to attempt to quantitate drug-likeness. Please give the code a try on your favorite molecules.









Hi Pat! Many thanks for giving MolSkill a try, we’re super happy that it’s getting attention from the community and generating useful discussion. We’d like to point out a few things in regards to the analyses you’ve run here:
ReplyDelete• MolSkill was specifically designed to distinguish compounds that are close to what “drug-like” molecules look like but that have features that for some reason are unattractive to the chemists’ eye.
• One of the things that we do emphasize in the paper, as well as in the repo, is to run the NIBR filters before running the default MolSkill model. This is because in practice, due to our sampling procedure and study design, the model has never seen a molecule that violates them. In fact, if we rerun the analyses that you kindly provided, but removing all compounds that do not feature a `CLEAR` flag in the NIBR filters, we can see that QED is no longer capable of distinguishing between the “odd” and “chembl_sample” datasets, whereas MolSkill can. (images in links below)
https://microsofteur-my.sharepoint.com/:i:/g/personal/jjimenezluna_microsoft_com/EcTtzMAM_ApMqxuT6D7JPNIBSvAPPrI4C4HnIcsDuWYcjw?e=utscfm
https://microsofteur-my.sharepoint.com/:i:/g/personal/jjimenezluna_microsoft_com/ETJMVPEPG31Chio9AQpY5n0BshhhLhDlbTgGUrgxQPrFDA?e=5d3DbA
A few of the molecules from the filtered “odd” set that are liked by QED but effectively deprioritized by MolSkill are presented below:
https://microsofteur-my.sharepoint.com/:i:/g/personal/jjimenezluna_microsoft_com/EXuPAzykTalOoVsT5kspZEABfqodXw9SJcidiyp4cFBq6g?e=4fgXPb
• Another aspect we’ve noticed is the drug molecule dataset contains structures that would be atypical of small molecule drug discovery projects (e.g., natural products, sugars, long aliphatic chains, steroids).
We do agree that MolSkill is somewhat of a black box, but we believe the fragment-based analyses provided in the manuscript help shed some light into what functional groups the model prefers. Additionally, one could either try and distill MolSkill scores via an inherently explainable model, or use feature attribution techniques as made popular by the explainable AI community. We have not explored either of these options.
All in all we’re very happy to see this highlighted in the blog and look forward to your feedback if you have further suggestions!
Thanks for your helpful feedback! I did notice the reference to the NIBR filters in the paper. I wanted to see what happened if I applied MolSkill the way I typically would on a drug discovery project.
ReplyDelete