We Need Better Benchmarks for Machine Learning in Drug Discovery

Most papers describing new methods for machine learning (ML) in drug discovery report some sort of benchmark comparing their algorithm and/or molecular representation with the current state of the art. In the past, I’ve written extensively about statistics and how methods should be compared. In this post, I’d like to focus instead on the datasets we use to benchmark and compare methods. Many papers I’ve read recently use the MoleculeNet dataset, released by the Pande group at Stanford in 2017, as the “standard” benchmark. This is a mistake. In this post, I’d like to use the MoleculeNet dataset to point out flaws in several widely used benchmarks. Beyond this, I’d like to propose some alternate strategies that could be used to improve benchmarking efforts and help the field to move forward. To begin, let’s examine the MoleculeNet benchmark, which to date, has been cited more than 1,800 times.
The MoleculeNet collection consists of 16 datasets divided into 4 categories.
- Quantum Mechanics – The datasets in this category contain 3D structures of molecules from the Generated Database (GDB) and associated properties that were calculated using quantum chemical methods. As I will discuss below, this dataset is frequently misused.
- Physical Chemistry - A set of measured values for aqueous solubility, free energy of solvation, and lipophilicity. In many respects, these benchmarks do not reflect how the underlying experimental measures are used in practice. I will expand on this in the sections below.
- Physiology - Datasets consisting of blood-brain barrier (BBB) penetration data and various toxicology readouts. The complexity of these endpoints makes the data less than ideal for benchmark comparisons.
- Biophysics - Five datasets that explore various aspects of protein-ligand binding. These datasets exhibit several problems, including assay artifacts, ambiguously defined chemical structures, and a lack of clear distribution between training and test sets. These points will be explored further below.
As I hope you’re beginning to see, the MoleculeNet dataset contains numerous flaws, making it difficult, if not impossible, to draw conclusions from method comparisons. I don’t want this post to be seen as an attack on the authors of MoleculeNet. I co-wrote a book with several of them and greatly respect their scientific abilities and accomplishments. However, if we want to move forward as a field, we need to establish high-quality benchmark datasets that will enable us to compare methods. We shouldn’t consider something a standard for the field simply because everyone blindly uses it. It should also be noted that while the points I make below primarily focus on MoleculeNet, other widely used benchmarks like the Therapeutic Data Commons (TDC) are equally flawed. In the sections below, I’ve outlined some criteria that I hope will start a conversation around benchmarking and what we need from datasets.
Technical Issues
We will begin with a few relatively simple topics surrounding data collection and reporting.
Valid Structures
A benchmark dataset should contain chemical structure representations that widely used Cheminformatics toolkits can parse. The BBB dataset in MoleculeNet contains 11 SMILES with uncharged tetravalent nitrogen atoms. This is not correct. A tetravalent nitrogen should always have a charge. As a result of these errors, popular Cheminformatics toolkits like the RDKit cannot parse the structures. I’ve seen dozens of papers that report results on this dataset, but I’ve yet to see anyone mention how these invalid SMILES were handled. Did the authors correct the SMILES and add the requisite positive charge? Did they skip these SMILES? Benchmark datasets should be carefully checked to ensure the validity of chemical structures.
Consistent Chemical Representation
The chemical structures in a benchmark dataset should be standardized according to an accepted convention. This is not the case with the MoleculeNet dataset. For example, consider the 59 beta-lactam antibiotics in the MoleculeNet BBB dataset. Depending on the structure, the carboxylic acid moiety in these molecules is represented in one of three different forms: the protonated acid, the anionic carboxylate, and the anionic salt form. We should compare the performance of molecular representations and ML algorithms rather than standardization methodologies.
Stereochemistry
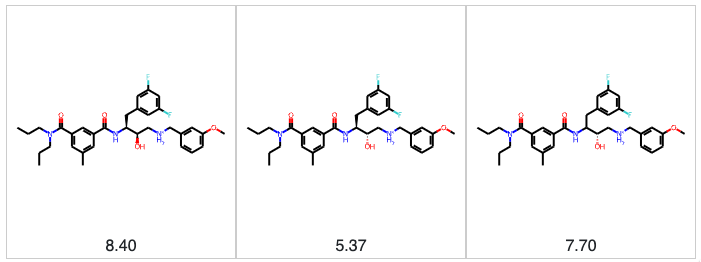
When modeling the relationship between chemical structure and some physical property or biological activity, we need to be confident that we are modeling the correct structure. In the section above, we talked about structures that cannot be parsed, but there are more subtle and nefarious structural issues we must deal with. Prime among these is stereochemistry. Stereoisomers can have vastly different properties and biological activities. As an example, let’s consider the BACE dataset in MoleculeNet. In the BACE dataset, we have 28 sets of stereoisomers. We have 3 stereoisomers in two cases; in the remaining cases, we have 2. The figure below shows one set of three stereoisomers. We have the 2R,3S stereoisomer with a pIC50 of 8.4 (4nM), and the 2S,3S stereoisomer that is 1,000-fold less potent, with a pIC50 of 5.37 (4µM). The structure on the right in the figure below is where things get dicier. The stereochemistry is defined for the hydroxyl group at the 2-position, but the difluorobenzyl group at the 3-position is racemic. At this point, we don’t know what we’re modeling. Is the racemic compound a 50/50 mixture of two stereoisomers, or does one stereoisomer predominate? We don’t know. This problem permeates the BACE dataset in MoleculeNet. 71% of the molecules have at least one undefined stereocenter, 222 molecules have 3 undefined stereocenters, and one molecule has a whopping 12 undefined stereocenters. Ideally, benchmark datasets should consist of achiral or chirally pure molecules with clearly defined stereocenters. It’s challenging to compare predictions when we don’t know the identities of the molecules being predicted.
Consistent Measurements
Ideally, the experiments used to define benchmark datasets should be conducted in the same lab under the same conditions. If benchmark datasets are aggregated from multiple labs, a set of standards should be used to ensure consistent results. This is certainly not the case for most of the datasets in the MoleculeNet collection. For example, the widely used MoleculeNet BACE dataset was collected from 55 papers. It is highly unlikely that the authors of these 55 papers employed the same experimental procedures to determine IC50s. Recent blog posts from Greg Landrum examined the impact of combining IC50 and Ki data from different assays. While Greg found that combining data from Ki assays is sometimes acceptable, the situation with IC50 data was more dire. Even under the best circumstances, 45% of values for the same molecule measured in two different papers differed by more than 0.3 logs, a value considered to represent the typical experimental error. Ultimately, if we combine datasets, we must do so carefully.
Realistic Dynamic Range and Cutoffs
The dynamic range of the data used in benchmarks should closely approximate the ranges one encounters in practice. This is not the case for the ESOL aqueous solubility dataset in MoleculeNet. Most pharmaceutical compounds tend to have solubilities somewhere between 1 and 500 µM. Assays are typically run within this relatively narrow range spanning 2.5 to 3 logs. Values less than 1µM are often reported as “<1µM” and values greater than 500 µM are reported as “>500µM”. This narrow dynamic range makes achieving good correlations between experimental and predicted values difficult. On the other hand, the ESOL dataset in MoleculeNet spans more than 13 logs, and it is easy to get a good correlation with very simple models. Unfortunately, this performance doesn’t reflect what one sees when predicting realistic test sets. For more information, please see this recent Practical Cheminformatics post.
On a related note, when evaluating classification models, one must set a cutoff value determining the boundary between molecules in one category and molecules in another. For example, consider the BACE dataset in MoleculeNet, which is used as a classification benchmark. Molecules with IC50 values less than 200nM are considered active, and molecules with IC50 values greater than or equal to 200nM are considered inactive. 200nM is an odd choice for an activity cutoff. It is quite a bit more potent than the values one finds with screening hits, which typically have IC50s in the single to double-digit µM range. The cutoff is also 10-20x greater than an IC50 one would target in lead optimization. Ultimately, the BACE classification benchmark doesn’t seem to reflect a situation we would encounter in practice.
Clear Definitions of Training, Validation, and Test Set Splits
When building a machine learning model, it’s important to divide the data into training, (sometimes) validation, and test sets. We want to avoid leaking information from the training set into the test set. In papers I read, there is no generally agreed-upon convention for dataset splitting. Some groups use random splits, others use various flavors of scaffold splitting, and some employ a cluster-based splitting strategy. To ensure consistency, it would be useful for each benchmark dataset to include labels indicating the preferred composition of training, validation, and test sets.
Data Curation Errors
In some cases, we encounter benchmark data that was incorrectly curated. As an example, consider the widely used BBB dataset in MoleculeNet. While this dataset has been used in hundreds of publications, no one appears to have noticed several serious errors. First, the dataset contains 59 duplicate structures. This is not the sort of thing we want to have in a benchmark. But wait, it gets worse, 10 of these duplicate structures have different labels. Yes, the BBB dataset has 10 pairs (see below) where the same molecule is labeled as both BBB penetrant and BBB non-penetrant. There are other errors such as glyburide being labeled as brain penetrant when the literature states the contrary. I’d like to thank my colleague Hakan Gunaydin for initially identifying some of these errors and sparking further investigation.
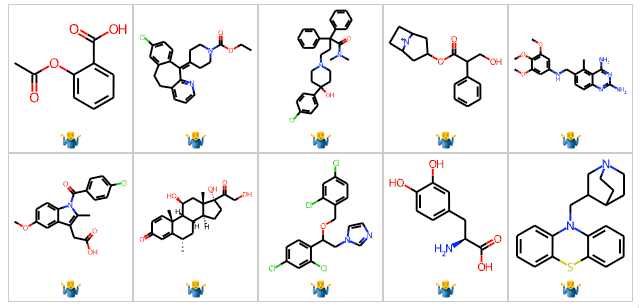
Understandably, errors can creep into these datasets. In many cases, people simply concatenate data tables from several papers and call that a benchmark dataset. We need to move to a more systematic approach where multiple groups evaluate benchmarks and confirm their validity.
Philosophical Issues
Moving on from purely technical matters, we transition to a more philosophical discussion of the characteristics that define a good benchmark dataset.
The Tasks Used in Benchmarks Should Be Relevant
Benchmark tasks should be relevant to typical work performed in a specific domain, in this case, drug discovery. Numerous papers have reported and compared results on the FreeSolv dataset in MoleculeNet. This dataset was designed to evaluate the ability of molecular dynamics simulations to estimate the free energy of solvation, which is an essential component of free energy calculations. However, this quantity, in and of itself, is not particularly useful, and I’ve never seen it used in isolation.
The MoleculeNet dataset also features the QM7, QM8, and QM9 datasets originally designed to evaluate the prediction of quantum chemical properties from 3D structures. It should be noted that most of these properties depend on the 3D coordinates of atoms and will vary with conformation. Unfortunately, this dataset has subsequently been misused, and many authors have reported and compared predictions of these properties from SMILES strings. While I appreciate that some claim a 1D representation like SMILES can capture some component of a 3D structure, predicting a property of one molecular conformation from a SMILES doesn’t make much sense.
Avoid Assays Having High Numbers of Artifacts
Another highly cited set from the MoleculeNet collection is the HIV dataset. This dataset consists of binary labels derived from 40,000 compounds tested in a cell assay designed to identify molecules that can inhibit HIV replication. Unfortunately, this dataset contains many compounds likely to generate assay artifacts. 70% of molecules labeled as “confirmed active” (CA) trigger one or more structural alerts. Of the 404 molecules labeled as CA, 68 are azo dyes widely known to be cytotoxic and generate assay interference. I wrote more about the problems with this dataset in a Practical Cheminformatics post in 2018. If we use a dataset as a benchmark, we should ensure that we are learning a true response and not an artifact.
For Now, Focus on Simple, Consistent, Well-Defined Endpoints
One of the most widely published MoleculeNet datasets is the blood-brain barrier penetration (BBB) set. This dataset contains 2050 molecules with a binary label indicating whether the molecule penetrates the BBB. Penetration of compounds into the central nervous system (CNS) is a complex process, and molecules can be actively transported or can take one of several paracellular routes through the BBB. The ability of a compound to enter the CNS can also be facilitated or hindered by certain disease states. Various experimental techniques have been applied to the measurement of CNS penetration. In some cases, this is based on animal studies; in other cases, measurements are taken from patients' cerebrospinal fluid (CSF). Many authors have simply classified any drugs used for psychiatric indications or drugs with side effects such as drowsiness as CNS penetrant. It should also be noted that experimental measurements of BBB penetration are widely variable. In a 2022 review by Haddad of the CNS penetration of antibiotics, the percentage of Ceftriaxone in CSF relative to plasma varied between 0.5 and 95%. Differences between free and protein-bound CNS concentrations further complicate this issue. While CNS penetration is important in numerous therapeutic areas, it’s not apparent that ML methods trained on flawed datasets significantly impact the field. We might benefit more from carefully studying consistently measured congeneric series where some compounds have shown CNS activity, and others have not. From the benchmarking standpoint, it’s difficult to compare methods based on a highly variable heterogeneous endpoint.
Considering poorly defined endpoints, we must discuss the toxicity-related datasets in MoleculeNet. There are 4 such datasets in the Physiology subset.
- SIDER - A collection of 1,427 chemical structures with associated side effects divided into categories ranging from “Hepatobiliary disorders” and “Infections and infestations” to less scientific categories such as “Product issues”, “Investigations”, and “Social circumstances”. Given the ambiguity and lack of mechanistic information associated with the side effects, this seems like a poor choice for a benchmark.
- Toxcast - A set of 8,595 chemical structures with 620 endpoints from a high-throughput screen using a panel of cell assays designed to assess a wide range of toxicities. This dataset is not a complete matrix; assay columns typically have ~2,500 binary labels and ~6,000 missing values. As might be expected, the dataset consists of many “bad actors”—56% of the molecules in the set trigger one or more structural alerts. Given the large number of assays, missing values, and ambiguities associated with HTS data, this seems to be a poor choice for a benchmark. Most papers comparing results for this dataset simply report an average classification area under the receiver operating characteristic (AUROC) for the 620 assays. This sort of comparison is uninformative and does little to demonstrate the utility of an ML model.
- Tox21 – This dataset is similar in spirit to the Toxcast dataset described above. It comprises 8014 compounds tested in 12 different quantitative HTS (qHTS) cell assays for nuclear receptor and stress response activity. As with Toxcast, the responses are binary, with between 189 and 961 active molecules per assay. The data here is less sparse than the Toxcast dataset, with between 575 and 2,104 missing values per assay. Of the four toxicity datasets, this one has the highest-quality data. However, the many complications associated with cell assays would not make these my first choice for benchmarking.
- Clintox – This dataset consists of 1483 SMILES strings and two binary labels indicating whether a molecule is an FDA-approved drug and whether a toxicity outcome has been reported. The molecules span the gamut from antibiotics to zinc oxide. Given the lack of specific information and the multitude of mechanisms for clinical toxicity observations, this seems like a poor choice for benchmarking predictive models.
It’s evident that toxicology prediction is important. We want to design drugs that are safe. Over the last 40 years, the pharmaceutical industry has developed a wide range of invitro and invivo assays for assessing the safety of drug candidates. In turn, computational groups have developed methods for predicting the outcomes of these assays based on chemical structure inputs. While it is often possible to establish relationships between chemical structure and a clearly defined endpoint, predicting complex, often subjective clinical outcomes based on chemical structures is problematic at best. Again, I’m not suggesting that the field should stop working on toxicity prediction, but I am saying that the MoleculeNet Tox datasets aren’t the best comparative benchmarks.
Some may argue that I’m advocating for the field to avoid working on difficult, important problems. Far from it. I’m simply asking that we walk before we try to run. For the most part, benchmarks have become bragging exercises. The authors put together a table showing how their method, highlighted in bold text, outperforms the current state of the art. Very little attention is given to why one method outperforms another or how one method does a better job highlighting particular chemical features. Perhaps a move to more robust, clearly defined benchmark datasets could help us to unravel what our models are doing and enable us to move forward as a field.
Next Steps
So far, I've talked a lot about about what's wrong. Let's change the focus to things we can do to improve the situation.
Where Do We Go From Here?
At this point, I hope I’ve convinced you that publishing benchmarks based on MoleculeNet is not a great idea. As I mentioned, the TDC dataset suffers from many of the same flaws as MoleculeNet and is an equally poor benchmarking tool. How should we move forward? I have a few ideas.
1. Focus benchmarking efforts on simple, robust, relevant, clearly defined endpoints. Initially, I would recommend aqueous solubility, membrane permeability, in vitro metabolic stability, and biochemical assays. These endpoints are reproducible, relatively inexpensive, and can provide reasonably large datasets. Until we develop a clear understanding of these benchmarks, we should avoid more complex cellular or in-vivo endpoints.
2. Develop a clear understanding of assay artifacts and introduce orthogonal measurements to identify “bad actors”. At the initial stage, this could be done with a panel of experienced practitioners, but we should move toward more objective measures.
3. Ensure that the chemical structures of the molecules in the dataset clearly represent the compounds used in an experiment. Focus experiments on achiral molecules and molecules with clearly defined stereochemistry. Ideally, it would also be beneficial to have evidence of compound purity and identity.
4. Capture and preserve the provenance of the data. Ensure that experimental procedures and experimental error measurements accompany datasets. Provide data showing the reproducibility of reference standards. This brings a couple of benefits. First, experimental reproducibility allows others to extend datasets or provide further evidence of experimental variability. Second, as we build ML models, we must clearly understand the relationships between model accuracy and experimental error.
5. Let’s use benchmarks to learn where our methods are working and where they aren’t. Simply flexing by putting your results in bold text in a table isn’t helping anyone. I realize that some people may not have the scientific background to interpret assay data, but this creates an excellent opportunity for collaboration with experimentalists.
6. Developing a benchmark is an ongoing activity. This point arose from an email exchange I had with Peter Eastman, one of the authors of the MoleculeNet benchmark. As Peter pointed out, version 1.0 of a benchmark will probably not be perfect. As people evaluate the benchmark, it needs to be updated and maintained. Hopefully, we can reach a point where benchmarks can be versioned, new data can be added, and the community can have more confidence in published results.
Where Will We Get These Datasets?
At this point, most readers are probably losing patience with me and asking two questions.
1. Just tell me which datasets I should use for benchmarking.
2. Given the draconian requirements above, where will we get these datasets?
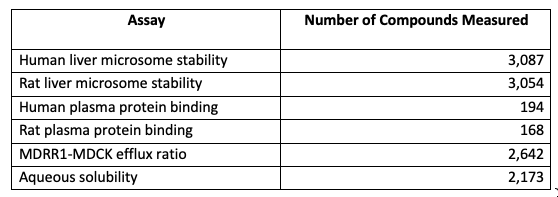
For question 1, I don’t currently have a lot of great answers. I’m scanning the literature and hope to have more suggestions soon. I’m also aware of ongoing efforts to curate quality datasets, but I don’t want to steal anyone’s thunder. As soon as these efforts are ready, I’ll eagerly publicize them. One dataset I like comes from a recent paper by Cheng Fang and coworkers at Biogen. In this paper, the authors provided a large dataset of ADME measurements on commercially available compounds similar to those found in drug discovery programs. All the structures and data are available in a machine-readable format on GitHub.
The Biogen dataset is a good start, but we need more data. Many have suggested that pharmaceutical companies should open more of their data to the community. It would be great to see more efforts like the one from Biogen, but intellectual property issues are at play here. When lawyers get involved, nothing moves quickly. In addition, each company runs its assays differently, and combining datasets will be difficult.
I think the best way to make progress on applications of machine learning to drug discovery is to fund a large public effort that will generate high-quality data and make this data available to the community. As a field, we don’t need another large language model that will produce incrementally better results on some meaningless benchmark. We need relevant, high-quality datasets that we can use as benchmarks. Beyond this, we must carefully evaluate the benchmark data to identify strengths and weaknesses in our methods and develop strategies to overcome limitations.
Over the last 10 years, there has been a substantial increase in the application of machine learning methods to a wide array of problems in drug discovery. Numerous academic computer science groups have entered the field and applied cutting-edge deep learning techniques to life science-related problems. While these efforts have a significant chance of advancing the field, the lack of appropriate benchmarks makes it challenging to determine whether we’re progressing. It frustrates me that funding agencies continue to support research into new ML methods but are unwilling to fund efforts to generate data that can be used to train and validate ML models for drug discovery. Much has been said about the potential of ML models to transform health care, but very little has been done to generate the data necessary to make this happen. I may be arrogant in saying this, but I think this data generation effort is one of the most important things we can do to impact human health.
As I said above, I don’t have all the answers, but I think we need to have a conversation. I’d love to talk with others who share similar or opposing views.
I Get By With A Lot Of Help From My Friends
Thanks to my many friends and colleagues for lively discussions on this important topic!


Timely post, Pat, and here are some thoughts on “Next Steps”.
ReplyDeleteFully agree that one should “focus benchmarking efforts on simple, robust, relevant, clearly defined endpoints”. I see benefits in balancing the representation of different chemotypes in data sets (I concede this is not so easy for IC50 data against individual targets) and having different measurements (PAMPA & MDCK permeability) for individual compounds. I’m sure that plenty of relevant data for dead Pharma projects are available although you’d probably need somebody on the inside to assemble the benchmark data sets for you. If I were still at AZ I’d be looking to assemble a matrix of IC50 values measured for reversible covalent (nitrile) Cathepsin inhibitors that had been synthesized in house (increases confidence that compounds are clean and that compounds are what is said on the bottles). Bear in mind that the dynamic range of Pharma anti-target assays (CYPs, hERG) is often limited because these are typically used for screening. In some cases (e.g. permeability assays) there is a non-obvious upper limit of quantitation that may differ between assays that appear to be very similar.
I agree that we need to have a clear understanding of nuisance behavior in assays and it’s important to be aware that interference with assay readout and undesirable mechanisms of action are distinct problems. With respect to nuisance behavior, it’s important to make a distinction between what we believe and what we actually know (compounds are frequently described as PAINS although I suspect that the number of articles on PAINS exceeds the number of compounds that have actually been shown to exhibit pan-assay interference).
Ha, I used the BBBP dataset in a tutorial one month ago, and the standardization workflow flagged those 11 SMILES, then some duplicates afterwards. I found it weird, but just removed everything without giving much thought to it. I'll try to use the Biogen datasets from now on.
ReplyDeleteYou make really relevant points as usual, and I'm glad that my conclusion was the same as yours: a project run by academia + pharma worldwide could fund the creation of a huge library with standardized measurements for relevant endpoints. There was that MELLODDY initiative, but they wouldn't disclose the structures. Literature datasets within the applicability domain of one's project could be used as test sets.