Free Wilson Analysis
How often have you been in this situation? You're working on a drug discovery project, you're in mid to late lead optimization, and you're wondering "what have we missed". "Are there promising combinations of substituents that we didn't synthesize"? Wouldn't it be great if you could look at all the substituent combinations that you didn't make, and identify the combinations that appear promising?

This work is licensed under a Creative Commons Attribution 4.0 International License.
Wow, you ask, what is this snazzy new technique? Actually, it's not a new technique, it's Free-Wilson analysis, which was originally published in 1964. The method is pretty simple and can be illustrated through an example like the one in a 1998 review by Hugo Kubinyi. In this blog post, I'll walk through the method using a Python implementation in GitHub.
1. R-group Decomposition
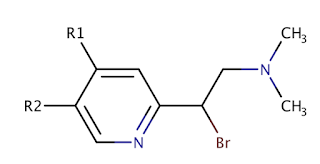
Let's assume we have a set of molecules built around this Markush structure.




1. R-group Decomposition
Let's assume we have a set of molecules built around this Markush structure.

In addition, let's assume that we have this set of molecules that vary in R1 and R2. The SMILES for these molecules are in the data directory in the GitHub repo.

The first step in performing the Free-Wilson Analysis is breaking the set of molecules down into a set of R-groups. This is done using the "free_wilson.py rgroup" command.
free_wilson.py rgroup --scaffold SCAFFOLD_MOLFILE --in INPUT_SMILES_FILE --prefix JOB_PREFIX
- SCAFFOLD_MOLFILE is a molfile for the scaffold with the substitution points labeled as R1, R2, etc.
- INPUT_SMILES_FILE is a SMILES file with the molecules. The input file doesn't have a header line and includes the SMILES and the molecule name. Here's a short example.
CN(C)CC(c1ccccc1)Br MOL0001
CN(C)CC(c1ccc(cc1)F)Br MOL0002
CN(C)CC(c1ccc(cc1)Cl)Br MOL0003
CN(C)CC(c1ccc(cc1)Br)Br MOL0004
CN(C)CC(c1ccc(cc1)I)Br MOL0005
- JOB_PREFIX is a prefix for the output file names. This can be anything you want.
For the example dataset in the GitHub repo, we can run something like this from the data directory.
../free_wilson.py rgroup --scaffold scaffold.mol --in fw_mols.smi --prefix test
Note that if you're on a Windows machine you may have to prefix all of the commands with "python". For instance:
python ../free_wilson.py rgroup --scaffold scaffold.mol --in fw_mols.smi --prefix test
At this point, you should see output like this.
Generating R-groups
wrote R-groups to test_rgroup.csv
R01: 6
R02: 6
wrote descriptors to test_vector.csv
The script tells you that there were 6 distinct R1 substituents and 6 distinct R2 substituents. In addition, the script writes out two files. The file test_rgroup.csv contains a breakdown of the R-groups for the molecules. This file mostly used for debugging and lets you know if the R-group decomposition worked as expected. I tend to use Vortex from Dotmatics to look at this kind of stuff. Here's what the file looks like in Vortex.

The second, more important, file is test_vector.csv. This file takes each molecule and converts it to a vector where each position in the vector represents a different R-group. In this case, the first 6 positions in the vector represent R1 (remember there were 6 distinct R1 substituents) and the next 6 positions represent the six different R2 groups.
Name,[H][*:1],F[*:1],Cl[*:1],Br[*:1],I[*:1],C[*:1],[H][*:2],F[*:2],Cl[*:2],Br[*:2],I[*:2],C[*:2]
MOL0001,1,0,0,0,0,0,1,0,0,0,0,0
MOL0002,1,0,0,0,0,0,0,1,0,0,0,0
MOL0003,1,0,0,0,0,0,0,0,1,0,0,0
MOL0004,1,0,0,0,0,0,0,0,0,1,0,0
MOL0005,1,0,0,0,0,0,0,0,0,0,1,0
MOL0006,1,0,0,0,0,0,0,0,0,0,0,1
MOL0007,0,1,0,0,0,0,1,0,0,0,0,0
The first line in the file is the header, which shows the substituents. A [*:1] following the substituent indicates R1, while [*:2] indicates R2. Graphically the vectors look like this.

2. Regression
Now that we have these vectors, we can use them to regress against the biological activity values and obtain coefficients the quantify the contributions of particular substituents to biological activity. We can do this with the "free_wilson.py regression" command.
free_wilson.py regression --desc DESCRIPTOR_FILE --act ACTIVITY_FILE --prefix JOB_PREFIX [--log]
- DESCRIPTOR_FILE is the vector description output from "free_wilson.py rgroup". This is test_vector.py above.
- ACTIVITY_FILE is a csv file with compound names and biological activity values. The header of this file should have the column names "Name" and "Act". The file should look like this.
Name,Act
MOL0001,7.46
MOL0002,8.16
MOL0003,8.68
MOL0004,8.89
MOL0005,9.25
MOL0006,9.30
- As above, JOB_PREFIX is the prefix for the output files.
- The final argument "--log" is an optional flag that indicates that the script should convert the activity values to a log scale. If, as above, the data is already log transformed, it is not necessary to use this flag.
For the example dataset in the GitHub repo, we can run something like this from the data directory.
../free_wilson.py regression --desc test_vector.csv --act fw_act.csv --prefix test
At this point, you should see output like this.
Running Ridge Regression
wrote model to test_lm.pkl
Intercept = 8.78
R**2 = 0.88
wrote comparison to test_comparison.csv
wrote coefficients to test_coefficients.csv
The script uses Ridge Regression to model the relationship between the vector derived from the R-groups and the activity values. The model is then serialized using the Python "pickle" function, and written out to a file, which will be used in the next step. Some summary statistics for the model are also output. The "free_wilson.py regression" command outputs two files. The first "test_comparison.csv" provides a comparison of the predicted and experimental values. I tend to use this file for debugging and identifying outliers. The second file, "test_coefficients.csv" provides the coefficients associated with each of the substituents. A positive coefficient indicates that the substituent increases the activity value, a negative coefficient indicates that the substituent decreases the activity value. The file looks like this.
R-Group SMILES,Coefficient,R-group,Count
[H][*:1],-0.135,1,6
F[*:1],-0.317,1,1
Cl[*:1],-0.039,1,4
Br[*:1],0.176,1,5
I[*:1],0.123,1,1
The first column is the substituent. As above, [*:1] after the substituent indicates R1, [*:2] indicates R2, etc. The second column indicates the coefficient, and the third indicates the substituent number (1 for R2, 2 for R2, etc). The final column indicates the number of times each substitent occurs in the dataset. I also tend to look at these files in Vortex, the R-group column becomes a checkbox in the Vortex filter device, making it easy to look at different R-groups. Here's an example of how this looks in Vortex.

Big substituents tend to increase the activity of the compounds, who knew?
In practice, I tend to run the program several times with different activity values and combine the results into a single table. This lets me get a more holistic view of how substituents affect different activities that we're trying to optimize. For instance, I might create a table showing the coefficients for cellular activity, hERG activity, and bioavailability. By looking at this combined view, I can get a better view of the strengths and liabilities of different substituents.
3. Enumeration
Finally, we'd like to be able to identify promising combinations of substituents that haven't been synthesized. This can be accomplished with the "free_wilson.py enumeration" command. This command generates all possible combinations of the available substituents. Please be aware that if you have several R-groups and a large number of possibilities for each R-group, this can generate a lot of molecules.
The "free_wilson.py enumeration" command works like this.
free_wilson.py enumeration --scaffold SCAFFOLD_MOLFILE --model MODEL_FILE --prefix JOB_PREFIX
- SCAFFOLD_MOLFILE is the scaffold molfile used in the first step above. This is necessary to generate the structures of the new molecules.
- MODEL_FILE is the pickled model that was generated in the second step above. This is necessary to generate the predicted activity of the new molecules.
- As above, JOB_PREFIX is used as a prefix for the output file names.
Note: As of 2018-09-14, I eliminated the --descriptor flag.
For the example dataset in the GitHub repo, we can run something like this from the data directory.
../free_wilson.py enumeration --model test_lm.pkl --prefix test --scaffold scaffold.mol
At this point, you should see output like this.
Enumerating New Products
Generated 14 products
wrote test_not_synthesized.csv
The output of this step is test_not_synthesized.csv, this file has the SMILES for the new products, the substituents, and the predicted activity It looks something like this.
SMILES,R1_SMILES,R2_SMILES,Pred
CN(C)CC(Br)c1ccc(F)c(F)c1,F[*:1],F[*:2],8.15
CN(C)CC(Br)c1ccc(Cl)c(F)c1,F[*:1],Cl[*:2],8.49
CN(C)CC(Br)c1ccc(Br)c(F)c1,F[*:1],Br[*:2],8.69
CN(C)CC(Br)c1ccc(I)c(F)c1,F[*:1],I[*:2],8.77
As above, I tend to run this for several different activity endpoints and integrate the data. This way, for instance, I could look for new molecules that would have good cell activity and bioavailability, while reducing hERG activity.
Doing This the Easy Way
I walked through this step by step to demonstrate how the program works, but the entire operation can be done in one step using the "free_wilson.py all" command.
free_wilson.py all --scaffold SCAFFOLD_MOLFILE --in INPUT_SMILES_FILE --prefix JOB_PREFIX --act ACTIVITY_FILE [--log]
The arguments are as discussed above so I won't go over them again.
For the example dataset in the GitHub repo, we can run something like this from the data directory.
../free_wilson.py all --scaffold scaffold.mol --in fw_mols.smi --act fw_act.csv --prefix test
The output is exactly the same as described above so I won't go through it again.
This work is licensed under a Creative Commons Attribution 4.0 International License.



Comments
Post a Comment