Using Reaction Transforms to Understand SAR
One of the most effective ways of understanding structure-activity relationships (SAR) is by comparing pairs of compounds which differ by a single, consistent feature. By doing this, we can often understand the impact of this feature on biological activity or physical properties. In order to effectively identify these pairs, we sometimes need an automated method that can sift through large datasets. In this post, we will show how we can use the chemical reaction capabilities in a Cheminformatics toolkit to identify interesting pairs of compounds. As usual, the code to accompany this post is on GitHub.
The idea of comparing the biological activity of pairs of compounds which only differ by a single feature has been a key concept since the beginning of medicinal chemistry. Over the last 15 years, software tools for generating "matched molecular pairs" (MMP) have become a common component of Cheminformatic analyses. For those unfamiliar with the technique, MMP analysis uses algorithms to identify pairs of molecules which differ by a single feature. For example, in the pairs below, the imidazopyridazine (IP) scaffold on the left has been replaced by a triazolopyridine (TP) scaffold on the right. Note that while the scaffold has changed, the substituents are the same.
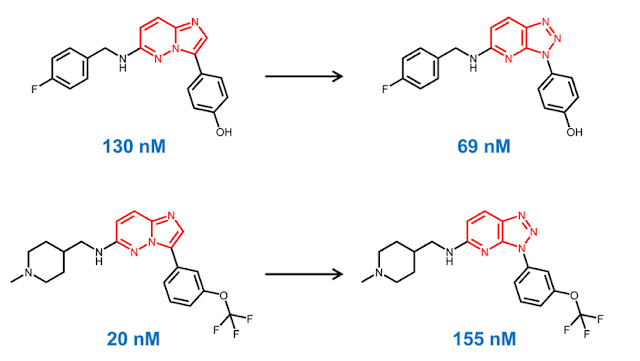
Let's say that we have a large dataset and we want to identify all of the pairs where the scaffold has changed from IP to TP, but everything else is the same.
We could run a standard matched molecular pair analysis, but there are two downsides to this.


The idea of comparing the biological activity of pairs of compounds which only differ by a single feature has been a key concept since the beginning of medicinal chemistry. Over the last 15 years, software tools for generating "matched molecular pairs" (MMP) have become a common component of Cheminformatic analyses. For those unfamiliar with the technique, MMP analysis uses algorithms to identify pairs of molecules which differ by a single feature. For example, in the pairs below, the imidazopyridazine (IP) scaffold on the left has been replaced by a triazolopyridine (TP) scaffold on the right. Note that while the scaffold has changed, the substituents are the same.

Let's say that we have a large dataset and we want to identify all of the pairs where the scaffold has changed from IP to TP, but everything else is the same.
We could run a standard matched molecular pair analysis, but there are two downsides to this.
- Many matched pair analysis methods are good at identifying changes in substituents, but sometimes fail to detect scaffold changes.
- Matched pair analysis techniques often identify dozens or even hundreds of pairs. We then have to sift through these pairs to find what we're looking for.

This reaction transform, which can be created in most chemical drawing programs (ChemDraw, Marvin Sketch, etc) defines the mapping between atoms in the reactant (left) and the product(right). With this reaction transform in hand, we can simply apply the transform to every molecule in our dataset. If the molecule contains the IP scaffold on the left, it will be transformed into the TP scaffold on the right. We can then check to see if the "product" molecule is in the original dataset. If it is, we have a pair. We can then collect these pairs and examine their differences in activity.
As an example let's consider this 2012 paper from Bioorganic & Medicinal Chemistry Letters, that discusses the hit to lead evaluation of a set of PIM kinase inhibitors. The ChEMBL database contains 57 structures from this paper, along with the corresponding PIM1 IC50 values. It would be useful to be able to determine the impact of the scaffold change on the PIM1 IC50. I've created a Git repository with the code for identifying the pairs, as well as a Jupyter notebook that demonstrates how to visualize the output.
To perform the analysis on the dataset in the repo, we can execute this command:
transformer_search.py --rxn rxn.rxn --in CHEMBL1949661.csv --out out.sdf
The output contains alternating pairs of molecules where the first contains the IP scaffold and the next contains the TP scaffold. The Jupyter notebook shows how we can then create a plot that compares the IC50 values for the two scaffolds. We can see that, for the most part, the IP scaffold is more active, but there are exceptions.

Of course, there's lot's more we could do, including making this plot interactive. We'll save that for another day. I'd like to thank Emanuele Perola for help in debugging the code and Greg Landrum and Lukas Pravda for sharing some RDKit tricks.



Comments
Post a Comment