IntroductionDataset splitting is one topic that doesn’t get enough attention when discussing machine learning (ML) in drug discovery. The data is typically divided into training and test sets when developing and evaluating an ML model. The model is trained on the training set, and its performance is assessed on the test set. If hyperparameter tuning is required, a validation set is also included. Teams often opt for a simple random split, arbitrarily assigning a portion of the dataset (usually 70-80%) as the training set and the rest (20-30%) as the test set. As many have pointed out, this basic splitting strategy often leads to an overly optimistic evaluation of the model's performance. With random splitting, it's common for the test set to contain molecules that closely resemble those in the training set.
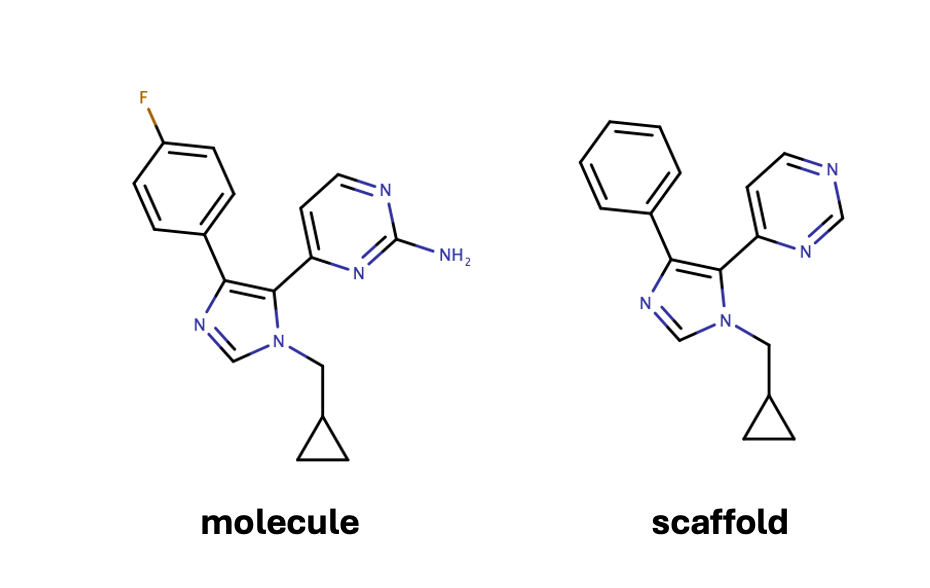
To address this issue, many groups have turned to scaffold splits. This splitting strategy, inspired by the work of Bemis and Murcko, reduces each molecule to a scaffold by iteratively removing monovalent atoms until none remain. This process simplifies the molecule while preserving its core structural features. For example, this method can reduce the molecule on the left to the scaffold on the right.
To split a dataset, unique scaffolds are identified, and each molecule is assigned a code based on its scaffold. The scaffold splitting strategy ensures molecules with the same scaffold are assigned to either the training or test set, but never both. However, one challenge with this approach arises when two highly similar molecules have distinct scaffolds.
For instance, consider molecule 1 and molecule 2 below, which differ by a single atom yet possess different scaffolds. Despite this difference, their Tanimoto similarity is 0.66, indicating high chemical similarity. It's reasonable to assume that the properties of these two molecules are comparable. If molecule 1 is included in the training set and molecule 2 in the test set, predicting the properties of molecule 2 becomes trivial.

In the real world, machine learning (ML) models are typically trained on historical data and then used to predict the properties or activities of molecules that will be synthesized in the future. Therefore, a time-based split of the data is ideal for evaluating the performance of an ML model. For example, if we had data for molecules tested over the course of a year, we could train a model on the molecules assayed between January and September and test it on the molecules assayed between October and December. However, most commonly used benchmark datasets don't have time stamps, making a time-based split impossible. While several groups have published methods for approximating a time-based split with an arbitrary dataset, none have gained widespread acceptance.
https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00787-9
https://arxiv.org/abs/2310.06399
Dataset Splitting Strategies for Drug Discovery
Despite considerable speculation and effort regarding dataset splitting, systematic comparisons have been scarce. This post introduces a framework for incorporating various dataset splitting techniques into cross-validation. I will utilize this framework to compare four popular splitting methods frequently employed when applying machine learning approaches in drug discovery.
Splitting Methods:
- Random Split: As previously mentioned, the data is randomly partitioned into training and test sets, with 80% allocated to the training set and the remaining 20% to the test set.
- Scaffold Split: Molecules are grouped based on their Bemis-Murcko scaffolds. This strategy ensures that molecules within the same cluster are assigned to either the training set or the test set.
- Butina Split: Morgan fingerprints are generated for the molecules, and the fingerprints are clustered using the Butina clustering implementation in the RDKit. Similar to the Scaffold Split, molecules in the same cluster are placed in either the training set or the test set.
- UMAP Split: Morgan fingerprints are generated for the molecules and projected into a two-dimensional space using the UMAP algorithm. Following a method proposed by Pedro Ballester's group, the UMAP coordinates are clustered into seven clusters using the agglomerative clustering implementation in scikit-learn.
This framework and comparison aim to provide clarity and guidance for selecting appropriate dataset splitting strategies for drug discovery applications. Throughout the following sections of this post, we will examine the variations in training and test set sizes resulting from different splitting strategies. Additionally, we will investigate the similarities between training and test sets and assess the impact of splitting on ML model performance. Appropriate statistical tests will be utilized to identify significant differences and areas where results converge. The code for dataset splitting and the analyses in this post is available on GitHub.
The Easy Way to Split a Dataset
It seems that the authors of every package for ML with molecules end up implementing their own methods for dataset splitting. This seems like a waste of time and often decreases the software's modularity. Ideally, we’d like to be able to plug and play with various molecular featurization methods and ML algorithms. Some groups have even implemented packages for specifically splitting chemical datasets. Again, I don’t think this is necessary.
The scikit-learn package has a method called
GroupKFold that allows the user to pass in a set of x variables (e.g., molecular descriptors), a set of y variables (e.g., IC50 values), and a set of groups (e.g., scaffold labels). This method then returns the indices of the training and test sets, where no examples from the same group are in both the training and test sets. Unfortunately, the default implementation of GroupKFold has a downside. It’s not possible to set the random seed. Using this method to perform cross-validation (CV), you’ll get the same splits for every CV fold. Fortunately, there is a workaround. I found a modified version called
GroupKFoldShuffle that allows the seed to be varied, making the method very useful in a CV context. I’ve incorporated this version of GroupKFoldShuffle into the useful_rdkit_utils package to make it easy to use. The application of GroupKFoldShuffle can best be explained through a simple code example, which I will explain below.
# 1. Read some data
df = pd.read_csv("/Users/pwalters/software/benchmark/data/biogen_logS.csv")
# 2. Add an RDKit molecule to the dataframe
df['mol'] = df.SMILES.apply(Chem.MolFromSmiles)
# 3. Instantiate a fingerprint generator
fpgen = rdFingerprintGenerator.GetMorganGenerator()
# 4. Generate fingerprints as descriptors
df['fp'] = df.mol.apply(fpgen.GetCountFingerprintAsNumPy)
# 5. Generate groups based on Bemis-Murcko scaffolds
df['bemis_murcko'] = uru.get_bemis_murcko_clusters(df.SMILES)
# 6. Instantiate a GroupKFoldShuffle object with 5 splits
group_kfold_shuffle = uru.GroupKFoldShuffle(n_splits=5, shuffle=True)
# 7. Perform cross-validation
for train, test in group_kfold_shuffle.split(np.stack(df.fp),df.logS,df.bemis_murcko):
print(len(train),len(test))
- We start by importing a dataset from a 2023 paper by the Biogen group.
- We need to add RDKit molecule objects to the dataframe to define molecular fingerprints as descriptors.
- Fingerprint generation requires an rdFingerprintGenerator object.
- Generate the fingerprints as numpy arrays.
- In this example, we will group the molecules using Bemis-Murcko scaffolds. We can use the get_bemis_murcko_clusters function from the useful_rdkit_utils package. This function takes a list of SMILES as inputs and returns a list of integers representing the scaffold associated with each molecule.
- Instantiate a GroupKFoldShuffle object and specify the number of splitting cycles and whether we want the data shuffled at each iteration.
- Use the GroupKFoldShuffle object to perform cross-validation. As we iterate over the CV folds, we provide the x and y variables and the groups. The returned values, train, and test contain the training and test sets indices, which can be easily transformed into dataframes using the Pandas iloc function.
The Impact of Dataset Splitting on Training and Test Set Sizes
Specific methods, such as GroupKFoldShuffle, utilize groups within a dataset to create partitions. These groups, often derived from clustering techniques, ensure that molecules from the same cluster are consistently assigned to the training or test set, preventing their inclusion in both. However, this approach can lead to training and test set size variations across multiple iterations. For instance, consider a dataset of 10 molecules divided into two clusters: cluster 1 with 6 molecules and cluster 2 with 4 molecules. Aiming for a 50/50 split into training and test sets, selecting cluster 1 as the training set in the first round of cross-validation would result in 6 molecules in the training set and 4 in the test set. Conversely, choosing cluster 2 as the training set in the second round would lead to 4 examples in the training set and 6 in the test set.
The example above may seem contrived. In real-world scenarios, how much do the sizes of training and test sets vary? To investigate this, I conducted five rounds of 5-fold cross-validation using the four splitting strategies mentioned earlier. For each fold, I noted the dimensions of the training and test sets and visualized the results in the box plot below. For those unfamiliar with box plots, I suggest visiting this Wikipedia page for further information.
The wide distribution of test set sizes for the UMAP clustering is odd, so I investigated further. The number of clusters (7) suggested by the
Ballester group seemed low, so I looked at test set size variability as a function of the number of clusters. As seen in the box plot below, the test set size becomes less variable when the number of clusters exceeds 35. As we move to a larger number of clusters, the size of the clusters and the subsequent test becomes more uniform. I don’t have a great answer for the ideal number of UMAP clusters yet. This aspect deserves further investigation.

Using Chemical Space Plots to Compare Dataset Splits
One common approach to comparing dataset splits is to plot the chemical space covered by training and test sets and color the points representing the training set one color and the test set a second color. The figure below provides examples of such a plot. In the plot, I’m using t-Distributed Stochastic Neighbor Embedding (tSNE) to project Morgan fingerprints in two dimensions for the Biogen solubility dataset mentioned above. In the plot, the training set is colored blue, and the test set is colored red. To be honest, I don’t find plots like this useful. The only obvious difference in the four plots below is with the umap_cluster plot, where the test set appears more “clumpy” than the other three. In addition to the fact that these plots aren’t quantitative, I also question the fidelity of the representation. The tSNE plots are projecting a 2048-dimensional representation into 2 dimensions. Knowing how faithfully the low-dimensional plot reproduces the distances in the high-dimensional space is difficult. In the next section, I’ll show what I think is a better way to represent the relationship between training and test sets.
Using Molecular Similarity to Compare Dataset Splits
In his
seminal 2004 paper, Bob Sheridan demonstrated that the similarity between training and test sets is a reliable predictor of ML model performance. Specifically, Bob's showed a strong correlation between model performance and the similarity of each test set molecule to its five nearest training set neighbors. Inspired by Bob's work, I created the plot below, which utilizes box plots to depict the distribution of similarities between the test set molecules and their five closest training set neighbors. The plots illustrate that random splits result in many test molecules similar to those in the training set. In contrast, Butina and scaffold splits yield fewer neighbors, while UMAP splits provide the smallest number of training set neighbors.
I have limited the previous plot to 10 cross-validation folds to save space and provide a more insightful visualization. Alternatively, we can gather all the cross-validation folds and display their combined distributions. The plot below presents the aggregate distributions for 5x5-fold cross-validation using the same Biogen solubility dataset. The trends in the aggregate plot mirror those observed in the previous plot. Random splits yield the most neighbors among the various splitting methods, followed by scaffold, Butina, and UMAP splits.
Although the boxplots above allow for a qualitative comparison of the distributions, determining whether the distributions differ significantly is crucial.
Tukey's Honestly Significant Difference (HSD) test offers a robust and convenient method for comparing all pairs of distributions while correcting for multiple comparisons. Notably, this test can be easily applied using just one line of code through the statsmodels Python library. The interested reader may want to consult these
Practical Cheminformatics posts for additional rants on statistical comparisons and ML models.
The output of the pairwise_tukeyhsd method in
statsmodels is a table like the one below.
Multiple Comparison of Means - Tukey HSD, FWER=0.05
=======================================================================
group1 group2 meandiff p-adj lower upper reject
-----------------------------------------------------------------------
butina_cluster random_cluster 0.0369 0.0 0.0357 0.0382 True
butina_cluster scaffold_cluster 0.0258 0.0 0.0246 0.0271 True
butina_cluster umap_cluster -0.0204 0.0 -0.0217 -0.0192 True
random_cluster scaffold_cluster -0.0111 0.0 -0.0124 -0.0099 True
random_cluster umap_cluster -0.0574 0.0 -0.0586 -0.0561 True
scaffold_cluster umap_cluster -0.0462 0.0 -0.0475 -0.045 True
The table compares the distribution means and indicates whether there is enough evidence to reject the null hypothesis, which assumes the means are equivalent at a significance level of 0.05.
group1 - the first group being compared
group 2 - the second group being compared
meandiff - the difference in the means of the distributions
p-adj - the adjusted p-value returned by the HSD test. In this case, a value < 0.05 is considered significant
lower - the lower confidence bound for the difference in the means
upper - the upper confidence bound for the difference in the means
reject - can we reject the null hypothesis that the means are the same?
In this case, we can reject the null hypothesis for all pairs of splitting methods. The pairwise_tukeyhsd method also has a nice visualization that shows the overlap between confidence intervals, providing a quick overview of the differences between methods. In this case, we can see that confidence intervals are small and don’t overlap between splitting methods.
Examining the Impact of Splitting Methods on ML Model Performance
Our discussion primarily focused on the test set size and its similarity to the training set. While these factors are important, our primary objective should be to evaluate the impact of data splitting on the predictive performance of our ML model. In this section, we’ll build an ML model and examine how splitting strategies impact performance. To simplify things, we’ll use Morgan count fingerprints from the RDKit as the molecular representation and LightGBM as the ML model. This representation and algorithm will perform 5 replicates of 5-fold cross-validation. To evaluate the performance of our ML model, we’ll use the coefficient of determination, R2.
The boxplot below compares the R2 values for 25 (5x5) ML models built using each splitting strategy outlined above. The results are consistent with what we observed when comparing the similarities of the training and test sets. Random splits with the highest train/test similarity generate the model with the highest R2, followed by scaffold, Butina, and UMAP splits.
We will again apply the Tukey HSD test to determine the statistical significance of the differences between the R2 distributions in the plot. The results of this test are presented in the table below.
Multiple Comparison of Means - Tukey HSD, FWER=0.05
========================================================
group1 group2 meandiff p-adj lower upper reject
--------------------------------------------------------
butina random 0.0523 0.0186 0.0065 0.0982 True
butina scaffold 0.0128 0.8838 -0.033 0.0587 False
butina umap -0.0558 0.0104 -0.1017 -0.01 True
random scaffold -0.0395 0.1167 -0.0854 0.0064 False
random umap -0.1082 0.0 -0.154 -0.0623 True
scaffold umap -0.0687 0.001 -0.1145 -0.0228 True
--------------------------------------------------------
In this case, we can reject the null hypothesis only for the comparisons between the UMAP and the other methods. This conclusion is also evident in the plot created by pairwise_tukeyhsd. The plot below showcases a useful feature of this method in statsmodels. Here, UMAP was set as the reference method when generating the plot. The dotted lines represent the upper and lower bounds for the mean of the UMAP distribution. The other methods are colored red, indicating the rejection of the null hypothesis.

Where Do We Go From Here?
This post briefly overviews some issues we encounter when splitting chemical datasets. I didn’t intend it to be comprehensive, and many open questions remain. I agree with the recent
paper from the Ballester group pointing out that scaffold splits provide an overly optimistic estimation of model performance. My current preference would be for Butina splits. While the UMAP splits appear more difficult, two factors concern me. First, the UMAP splits use a projection that only partially captures the similarity between the chemical structures in the dataset. Second, agglomerative clustering tends to generate clusters of very different sizes, which could artificially inflate the variance in the metrics used for prediction.
In this post, I focused on a single dataset. It’s possible that some of what we observed was a function of the dataset we used. With a more congeneric dataset, we could arrive at different conclusions. I could continue this post for another five pages and provide a complete analysis of several datasets. However, that’s probably not what any of us want. A Jupyter notebook with the code is available for those interested in additional analyses. In fact, the notebook has a link to another dataset that is very different from the dataset we just examined. I’d urge interested readers to try the code and see how the methods perform on their datasets. As always, I’m open to discussions. Please reach out if you’d like to discuss or have ideas for other ways to split datasets or compare splitting strategies.










Nice article, Pat. I always read articles. I just want to share that apart from random splitting, there are Sphere Exclusion, D-Optimal (Fedorov), CADEX and few other statistical methods for data splitting for regression problem.
ReplyDeleteFor classification problem, in addition, we have stratified Sphere exclusion, stratified random, Stratified D-Optimal (Fedorov), stratified CADEX and also stratified maximum dissimilarity using Tanimoto.
These methods are not only useful to avoid bias but also minimize purpose selection of the training set and the test set. As, purpose selection could lead to 'Information leakage', other methods scaffold, etc. should be used cautiously.
Thanks, the Butina method is similar in spirit to sphere exclusion. I appreciate the pointers to other methods.
DeleteNice work Pat, this very much reflects what we have often found with regards to train/test splits. One thing we have recently done, more like an asymptotic extreme case, is to iteratively optimise the separation between train and test splits. This is done by a simple stochastic optimisation described here https://github.com/IanAWatson/LillyMol/blob/master/docs/GFP/train_test_split_optimise.md.
ReplyDeleteIt usually quickly finds well separated train and test splits, significantly more separated than what other methods find.
I just took the Biogen data you used here and applied the splitting optimisation to the Biogen datasets and then build models on those splits. I find models worse than what you find with the splits above -
https://github.com/IanAWatson/LillyMol/blob/master/docs/Workflows/train_test_split_optimise.md
indicating likely further separation between train and test.
But rather than what might be expected as a real-world kind of split, the optimised splitting is more of a worst case scenario.
But there is a fairly straightforward way of generating very strongly separated train/test splits if anyone wants to determine something approximating a worst case scenario.
Ian
Thank you for sharing your results on another dataset and your insights in this informative post.
ReplyDeleteIn our preprint (now published at https://link.springer.com/chapter/10.1007/978-3-031-72359-9_5), we compared test set performance using scaffold split, Butina split, and UMAP split on the same dataset. This analysis was conducted across 60 cancer-drug-response datasets using three machine learning methods. Our findings supported the hypothesis that scaffold split overestimates virtual screening performance. Importantly, we also demonstrated that selecting models based on scaffold-split test results is likely to lead to suboptimal prospective outcomes.
We recently submitted an invited extension of this paper, which is currently under review. This extension adds the random split to our comparisons and further discusses the advantages of using the UMAP split. Qianrong Guo’s code (https://github.com/ScaffoldSplitsOverestimateVS/ScaffoldSplitsOverestimateVS) can be adapted to perform similar analyses with these four splits on other datasets.
It is interesting that the conclusions with this new dataset are essentially the same: avoid the scaffold split, and recognize UMAP split as the most challenging. These results further support the generality of this approach. Have you encountered any dataset where this is not the case?
You also outline your reasons for currently preferring Butina over UMAP, namely:
1. UMAP may not perfectly capture the distance between molecules.
2. Variability in UMAP test set sizes increases variability in performance metrics across left-out clusters.
These are valid points, but I would like to offer a few considerations.
Regarding point 1. In a previous study (https://www.mdpi.com/2218-273X/13/3/498), we compared the clustering quality of Butina and UMAP, showing that UMAP clustering is superior, at least in these 60 datasets. Butina clustering has known issues, such as classifying points very close to a cluster as outliers. These issues thus seem to outweigh UMAP’s limitations. Furthermore, we observed that Butina splits are substantially easier than UMAP splits across all 60 datasets. For instance, the best method achieved a submicromolar hit rate of 12% with the UMAP split but 46% with the Butina split. From our prospective results on two unrelated cancer cell lines (unpublished), the method can indeed discover novel cell-active submicromolar inhibitors, although with a hit rate lower than that of the UMAP split.
Regarding point 2: For our datasets, K=7 was the optimal number of clusters. You can determine the optimal K for your dataset using Saiveth Hernandez’s code (https://github.com/Sahet11/nci60_clustering). There are typically many large K values that yield near-optimal clustering quality, which can hence reduce test size variability if used. Also, I would expect that selecting a K tailored to your dataset would result in an even more challenging UMAP split.