Using Reaction Transforms to Understand SAR
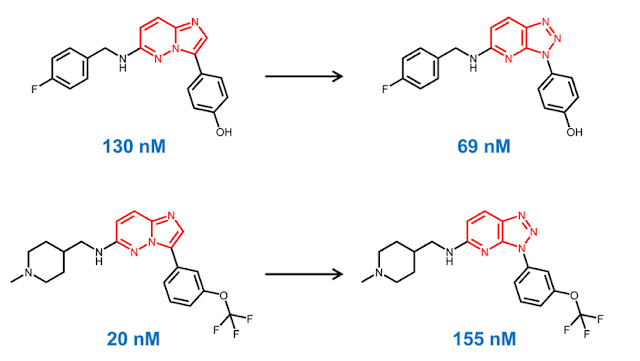
One of the most effective ways of understanding structure-activity relationships (SAR) is by comparing pairs of compounds which differ by a single, consistent feature. By doing this, we can often understand the impact of this feature on biological activity or physical properties. In order to effectively identify these pairs, we sometimes need an automated method that can sift through large datasets. In this post, we will show how we can use the chemical reaction capabilities in a Cheminformatics toolkit to identify interesting pairs of compounds. As usual, the code to accompany this post is on GitHub . The idea of comparing the biological activity of pairs of compounds which only differ by a single feature has been a key concept since the beginning of medicinal chemistry. Over the last 15 years, software tools for generating " matched molecular pairs " (MMP) have become a common component of Cheminformatic analyses. For those unfamiliar with t...